数据库与数据格式
Updated on: 2024-03-15 08:26:04
一、短序列数据库
1.INSDC : International Nucleotide Sequence Database Collaboration (insdc.org)
由SRA(NCBI的短序列片段数据库)、ENA(欧洲EBI的短序列片段数据库)和DAR(日本DDBJ的短序列片段数据库)合作的组织,三者之间共享数据。
2.CLIPZ : http://www.clipz.unibas.ch
交联免疫共沉淀实验与RNA结合蛋白获得的短序列片段数据库,支持注释和可视化以及数据挖掘与分析
3.NGSmethDB :NGSmethDB - Database Commons (cncb.ac.cn)
收录了人类、鼠和拟南芥和两种胞嘧啶甲基化数据。
二、数据格式

2.1序列及质量分数格式
①FASTA格式
FASTA 格式是一种基于ASCII 码的文本的格式,可以存储一个或多个核苷酸序列或肽序列数据。在FASTA格式中,每一个序列数据以单行描述开始(必须单行),后跟紧跟一行或多行序列数据。下一个序列数据也是如此,循环往复。
FASTA 格式文件中的每个序列信息由两个部分组成:
1. 描述行 (The description line, Defline, Header or Identifier line): 以一个大于号(“>”)开头,内容可以随意,但不能有重复,相当于身份识别信息。
2. 序列行 (Sequence Line):一行或多行的核苷酸序列或肽序列,其中碱基对或氨基酸使用单字母代码表示。

注意:在FASTA 格式文件中,内容中间不允许有空行,建议所有文本行的长度小于 80 个字符,序列超过80个字符的部分紧跟着换到下一行。
②CSFASTA格式
与FASTA格式类似,只不过序列是颜色编码的,即color-space的。我认为这种格式其实对应与SoLiD测序方法,应该是这种方法产生的数据格式。
③FASTQ格式
使用最多的一种文本格式,包含了测序片段的序列和对应的质量分数。
下面是一个Illumina平台测序的真实数据,其中包含了1条reads的信息。
@ST-E00126:128:HJFLHCCXX:2:1101:7405:1133
TTGCAAAAAATTTCTCTCATTCTGTAGGTTGCCTGTTCACTCTGATGATAGTTTGTTTTGG
+
FFKKKFKKFKF<KK<F,AFKKKKK7FFK77<FKK,<F7K,,7AF<FF7FKK7AA,7<FA,,FASTQ格式储存的序列信息,每1条reads的信息,可以分成4行。
第1行主要储存序列测序时的坐标等信息
@ST-E00126:128:HJFLHCCXX:2:1101:7405:1133@ 开始的标记符号
ST-E00126:128:HJFLHCCXX 测序仪唯一的设备名称
2 lane的编号
1101 tail的坐标
7405 在tail中的X坐标
1133 在tail中的Y坐标
第2行是测序得到的序列信息,一般用ATCGN来表示,其中N表示荧光信号干扰无法判断到底是哪个碱基。
第3行以“+”开始,可以储存一些附加信息,一般是空的。
第4行储存的是质量信息,与第2行的碱基序列是一一对应的,其中的每一个符号对应的ASCII值成为phred值,可以简单理解为对应位置碱基的质量值,越大说明测序的质量越好。不同的版本对应的不同。
详细谈谈FASTQ质量值的计算方法
在测序仪进行测序的时候,会自动根据荧光信号的强弱给出一个参考的测序错误概率(error probility,P)根据定义来说,P值肯定是越小越好。我们怎么储存他们呢?直接储存成小数点?比如1%储存成0.01?这肯定是不高效的,因为1个碱基的信息,占用了至少4个字符。
所以科学家们的做法想了一个办法:
1.将P取log10之后再乘以-10,得到的结果为Q。
比如,P=1%,那么对应的Q=-10*log10(0.01)=20
2.把这个Q加上33或者64转成一个新的数值,称为Phred,最后把Phred对应的ASCII字符对应到这个碱基。
如Q=20,Phred = 20 + 33 = 53,对应的符号是”5”
这样就可以用1个符号与1个碱基一一对应,是不是很聪明?!
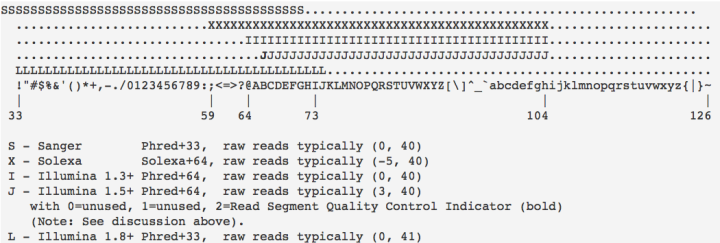
各版本不同Phred对应的ASCII值
* 在计算Q值和加上33/64的时候,不同测序仪,产生的数据不同,大概如下所示:
Solexa标准
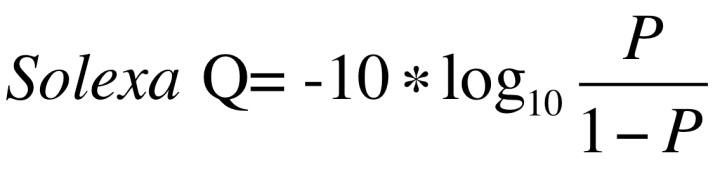
Illumina标准
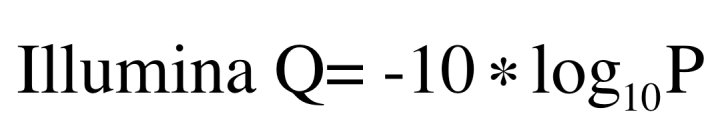
不同测序仪的不同Phred值对应的ASCII表
Solex: Q+64取ASCⅡ码
Illumina: Q+33取ASCⅡ码
当然两者的Q算法也不同
④CSFASTQ格式
同CSFASTA,不过类似于FASTQ。
⑤Qseq和SCARF
是Illumina平台自己的数据格式。
Qseq类似FASTQ,保存了测序仪名称、测序编号、条带编号、坐标等,每行保留一条序列信息,以“.”保存未知碱基(N)。并且其质量分数的计算也与FASTQ不同是pherd +64取ASCⅡ码。
SCARF类似,只是质量分数的计算采用了Solexa的方式。
⑥一些二进制类型格式
二进制类型格式一方面能够减小存储大小,但是也严重减少了阅读与修改的能力。
⑦小结

2.2比对数据格式
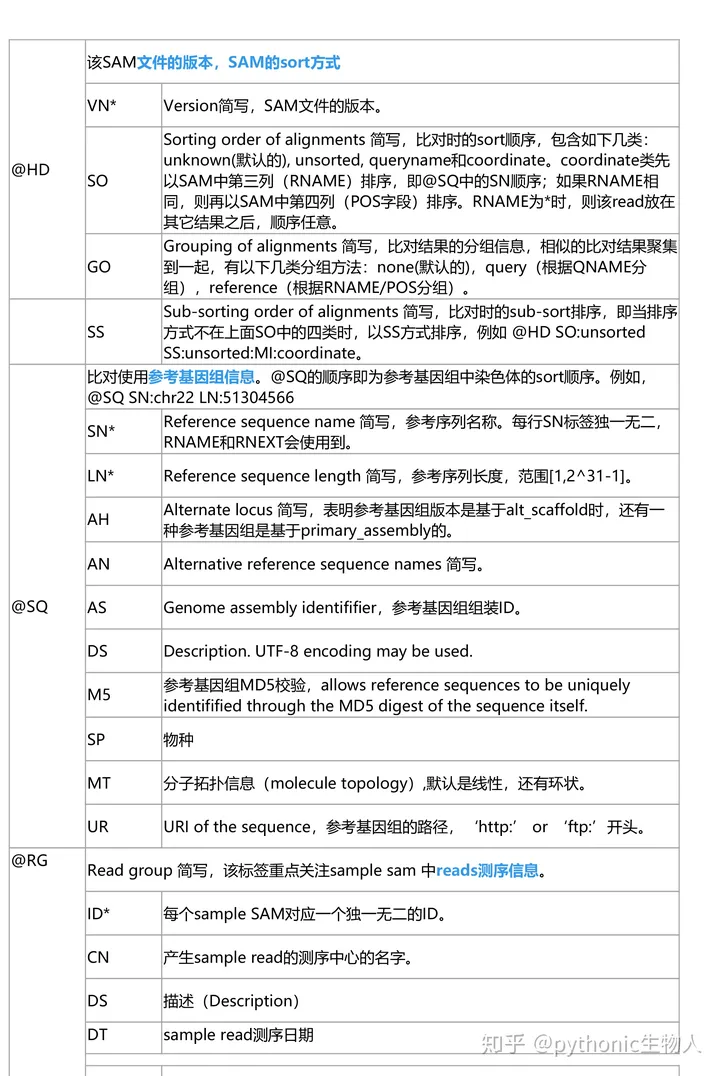
①SAM格式
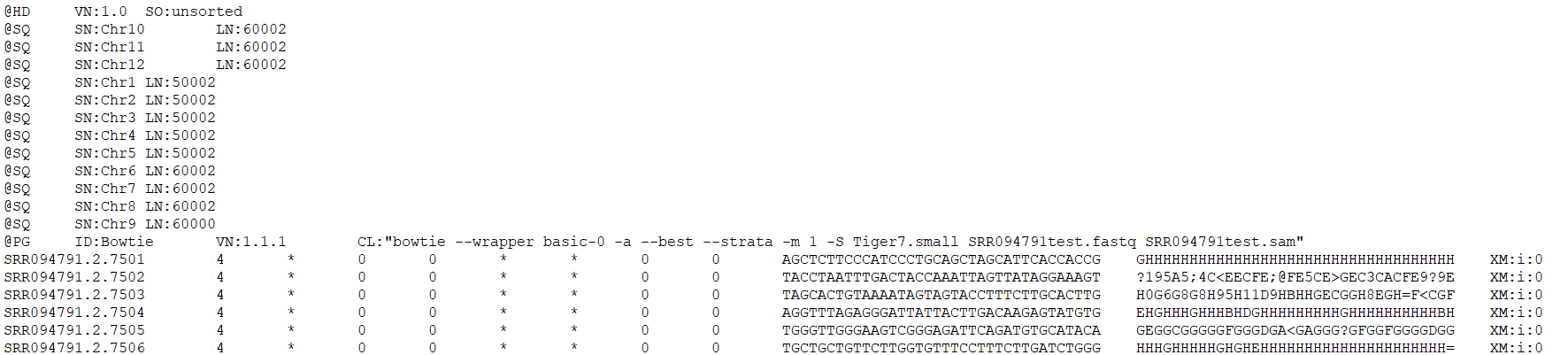
sam格式是最常见的保存比对数据的文件格式,通常包含两部分:头部分和比对部分

a. 标头部分解释
b. 比对部分
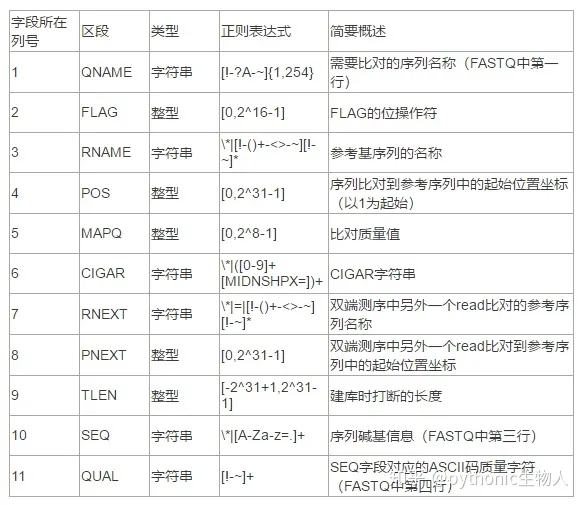
最重要的是前8个flag ,这里采用2进制的方式进行存储Flag,并且满足对应情况时该位为1,不满足时该位为0,最后转为唯一的一个十进制数,表示这条read对比的情况如何。*表示缺省。
1 :该read使用双端测序,单端测序为0;
2: 该read和完全比对到参考序列;
4: 该read没有比对到参考序列;
8: 双端序列的另外一条序列没有比对上参考序列(read1或者read2);
16:该read比对到参考序列的负链上(该read反向互补比对到参考序列);
32 :该read的另一条read比对到参考序列的负链上;
64 :双端测序 read1;
128 : 双端测序read2;
256: 该read不是最佳的比对序列,一条read能比对到参考序列的多个位置,只有一个是最佳的比对位置,其他都是次要的;
512: 该read在过滤(碱基质量,测序平台等指标)时没通过;
1024: PCR(文库构建时)或者仪器(测序时)导致的重复序列;
2048: 该read可能存在嵌合(发生在PCR过程中),当前比对部分只是read的一部分;再详细说说第5列MAPQ,即比对的质量:
对应参考序列的质量(MAPing Quality),比对的质量分数,越高说明该read比对到参考基因组上的位置越准确。其值等于-10 lg Probility (错配概率),得出值后四舍五入的整数就是MAPQ值。如果该值是255,则说明对应质量无效。例如,MAPQ为20,即Q20,错误率为0.01,20 = -10log10(0.01) = -10*(-2)。
② bam格式
为了便于调用,传输和存储而将sam文件转为二进制获得的文件,其读取更为迅速,占用空间更小。并且能够生成索引提高读取速度。
③ 其他不常见格式
| 名称 | 类型 | 描述 | 后缀 |
|---|---|---|---|
| PSL | 文本 | BLAT程序产生的比对格式 | .psl |
| bowtie | 文本 | Bowtie产生的默认格式 | .bowtie |
| maq | 二进制 | Maq产生的格式 | .map |
2.3 序列组装格式
①ACE格式
应用最广的组装格式,存储了contig和read信息还有序列、质量分数、起止坐标等,还提供可视化
②CAF格式
最初为了有效存储所有的数据,使所有组装程序以此为接口想结合而设计,因此更具兼容。在Sanger测序法中使用更多。
2.4注释及可视化信息格式
①BED格式
BED格式可用于UCSC的Genome Browser可视化工具中。同时主要是用于对基因组的特征或注释信息。
BED格式每列表示不同的信息,用制表符分开:
1. chrom - 染色体名称 (e.g. chr3, chrY, chr2_random) 或 scaffold (e.g. scaffold10671).
2. chromStart - 染色体或scaffold起始位点. 注意!是从0开始计数!是从0开始计数!是从0开始计数!
3. chromEnd - 染色体或scaffold终止位点. 注意!结束位点的碱基不包含在内。比如chromStart=0, chromEnd=100, 那么实际上表示为 0-99.
4. name - BED的名称.
5. score - 分数为0 到1000。颜色越深,分数越高。
6. strand - 链方向. "." (=no strand) 或"+" 或 "-".
7. thickStart - 绘制特征的起始位置(例如起始密码子)。当没有thick部分时,thickStart和thickEnd通常设置为chromStart位置。
8. thickEnd - 绘制特征的终止位置(例如终止密码子)。
9. itemRgb - RGB值 (e.g. 255,0,0). 如果track 行 itemRgb 参数设置为 "On", RBG值会决定BED文件中该行数据的颜色.
10. blockCount - blocks (exons)在BED line的数量.
11. blockSizes - 用逗号分隔的列表,用来表示block的的大小。列表中数字的数目应和blockCount 一致。
12. blockStarts - 用逗号分隔的列表,用来表示block的起始。 所有blockStart 的位点应该和chromStart 相关。 列表中数字的数目应 和blockCount 一致。在有block等相关列的BED文件中,blockStart* 值必须是 0, 因此第一个block是从*chromStart* 开始。同样的,最后一个blockStart* 位点加上最后一个blockSize 值应该和chromEnd相等。Blocks不能重叠.
BED格式样例,有几列就叫BED几,例如BED9有上述顺序数来9列的信息
track name="ItemRGBDemo" description="Item RGB demonstration" itemRgb="On"
chr7 127471196 127472363 Pos1 0 + 127471196 127472363 255,0,0
chr7 127472363 127473530 Pos2 0 + 127472363 127473530 255,0,0
chr7 127473530 127474697 Pos3 0 + 127473530 127474697 255,0,0
chr7 127474697 127475864 Pos4 0 + 127474697 127475864 255,0,0
chr7 127475864 127477031 Neg1 0 - 127475864 127477031 0,0,255
chr7 127477031 127478198 Neg2 0 - 127477031 127478198 0,0,255
chr7 127478198 127479365 Neg3 0 - 127478198 127479365 0,0,255
chr7 127479365 127480532 Pos5 0 + 127479365 127480532 255,0,0
chr7 127480532 127481699 Neg4 0 - 127480532 127481699 0,0,25②BIGBED
二进制压缩版的BED或bedgraph,压缩方式是将前三列的位置信息用二进制的索引代替。
转换时可以使用bedToBigBed 和 bigBedToBed 互相转换
除了上述两种bed文件格式,还有bedGraph beddetail。
③wig(wiggle)和bigwig
④GTF和GFF
目前只尝试过使用GFF3格式文件在IGV视图上进行可视化。