二代测序方式及原理
Updated on: 2024-02-28 19:44:10
一、illumina测序
1. 基本流程
1.1样本建库:
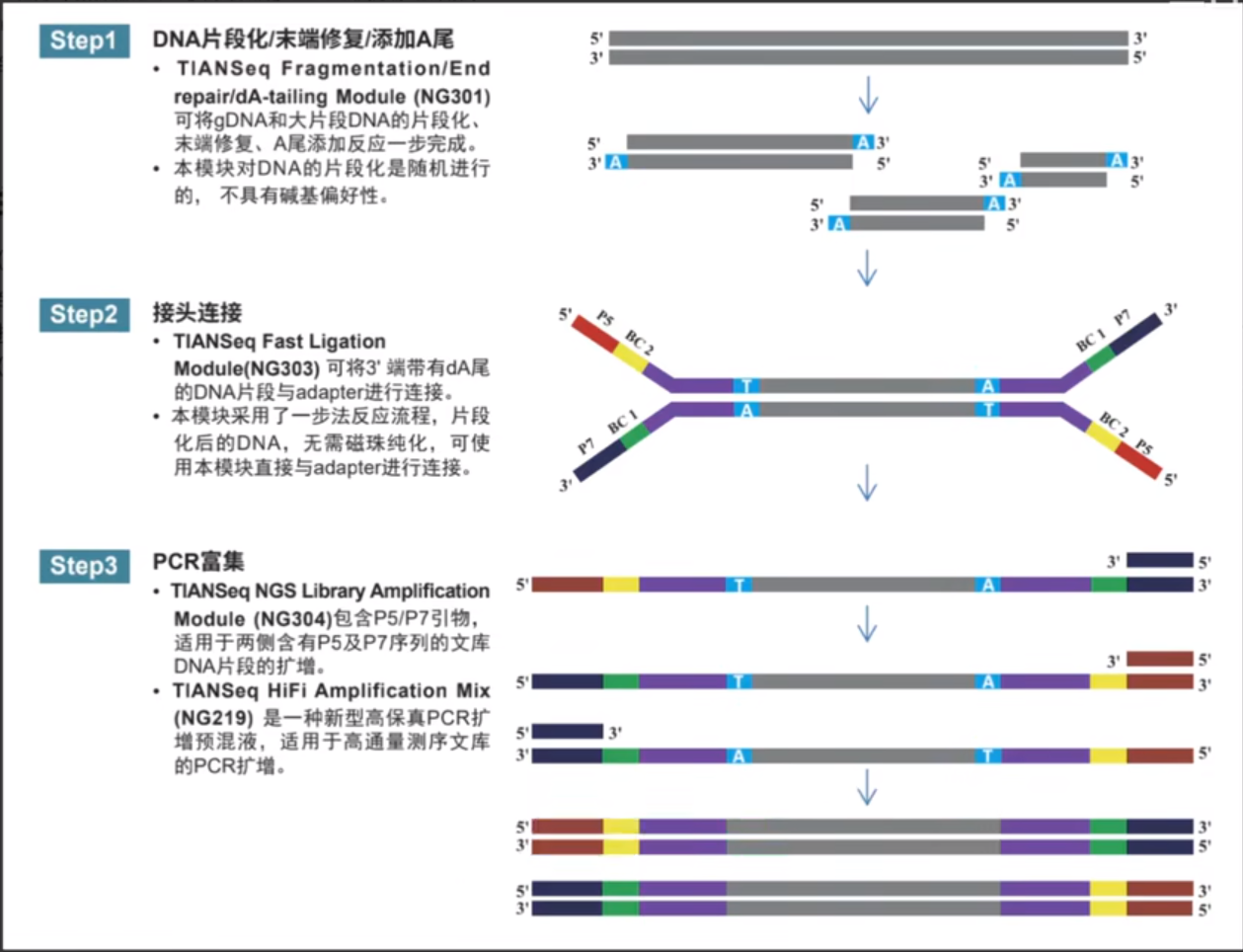
①将提取的DNA通过超声波(或雾化,酶切)随机打碎成小片段,随后用DNA聚合酶补齐并添加A尾
②通过A尾将接头接上,其中接头包括三个部分:最内端的测序结合位点(紫色)用于边合成边测序,中间的BarCode(黄色和绿色)也称为index,一般是6个bp,用于区分样品来源,最外端的扩增引物段(红色和黑色)用于step3的PCR扩增
1.2簇生成
①flowcell结构:一个flowcell包含4~10个lane(依据测序方式不同,常为6或8个),lane两端含有小孔可以使液体流入流出;lane含有2行swath,每个swath含有多个小格子,测序反应在小格子内进行。整个flowcell大小同载玻片相似。
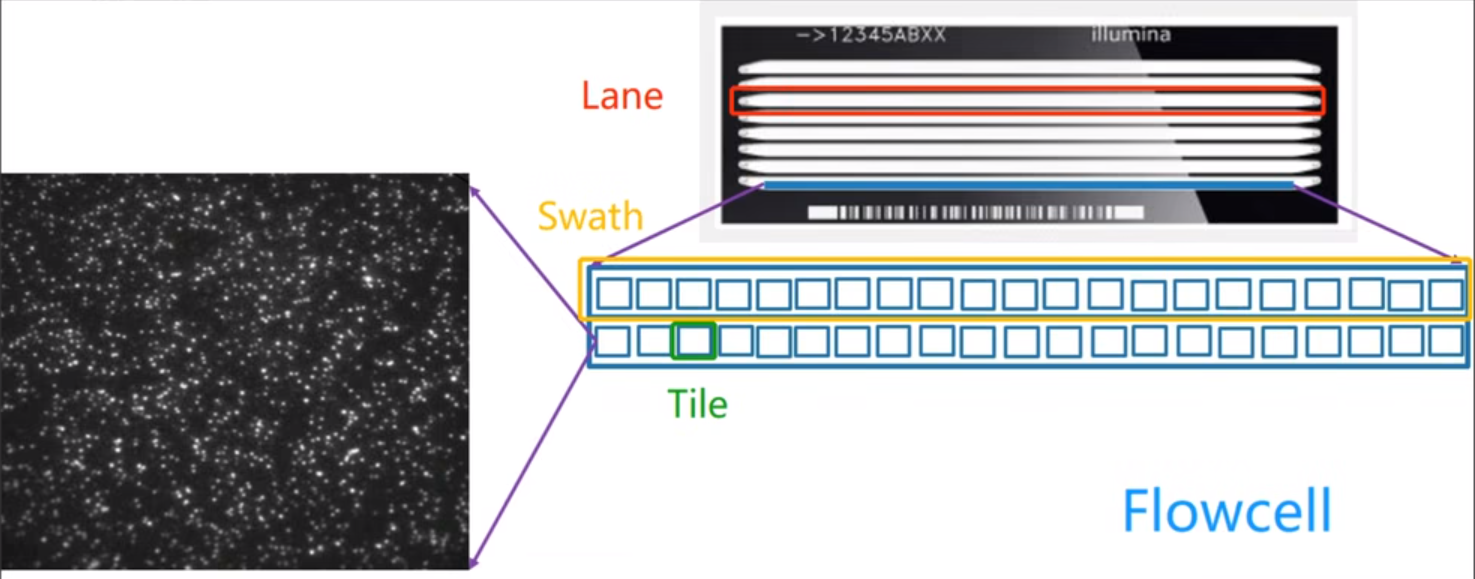
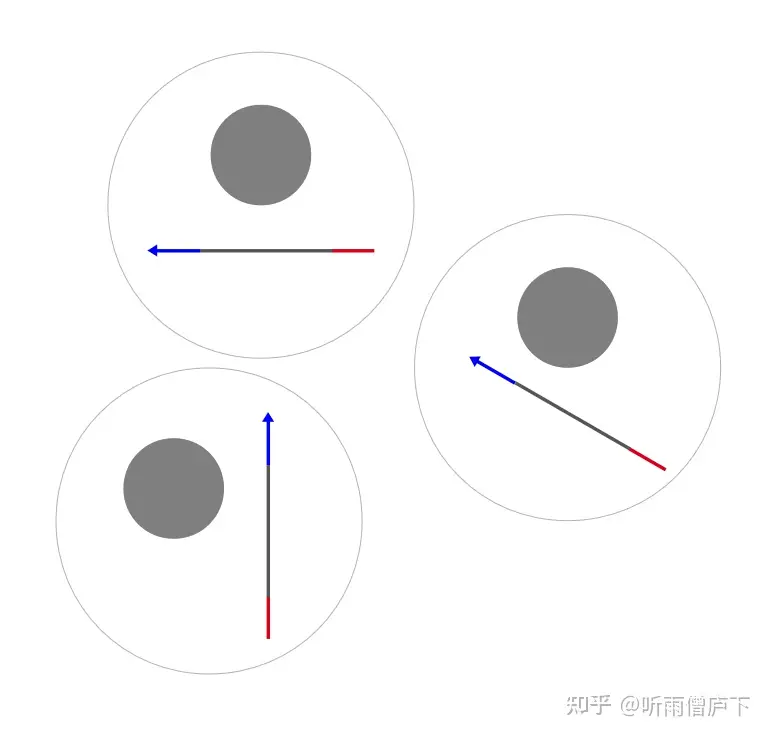
②lane内部含有两种通过共价键被直立固定在表面的寡核苷酸链(紫色和蓝色),这些寡核苷酸链与建库时添加的接头最外端序列(P5和P7)互补,充当引物的作用。

③需要测序的模板链接头与对应的固定片段杂交,此时DNA聚合酶可以以被测序链为模板进行延伸
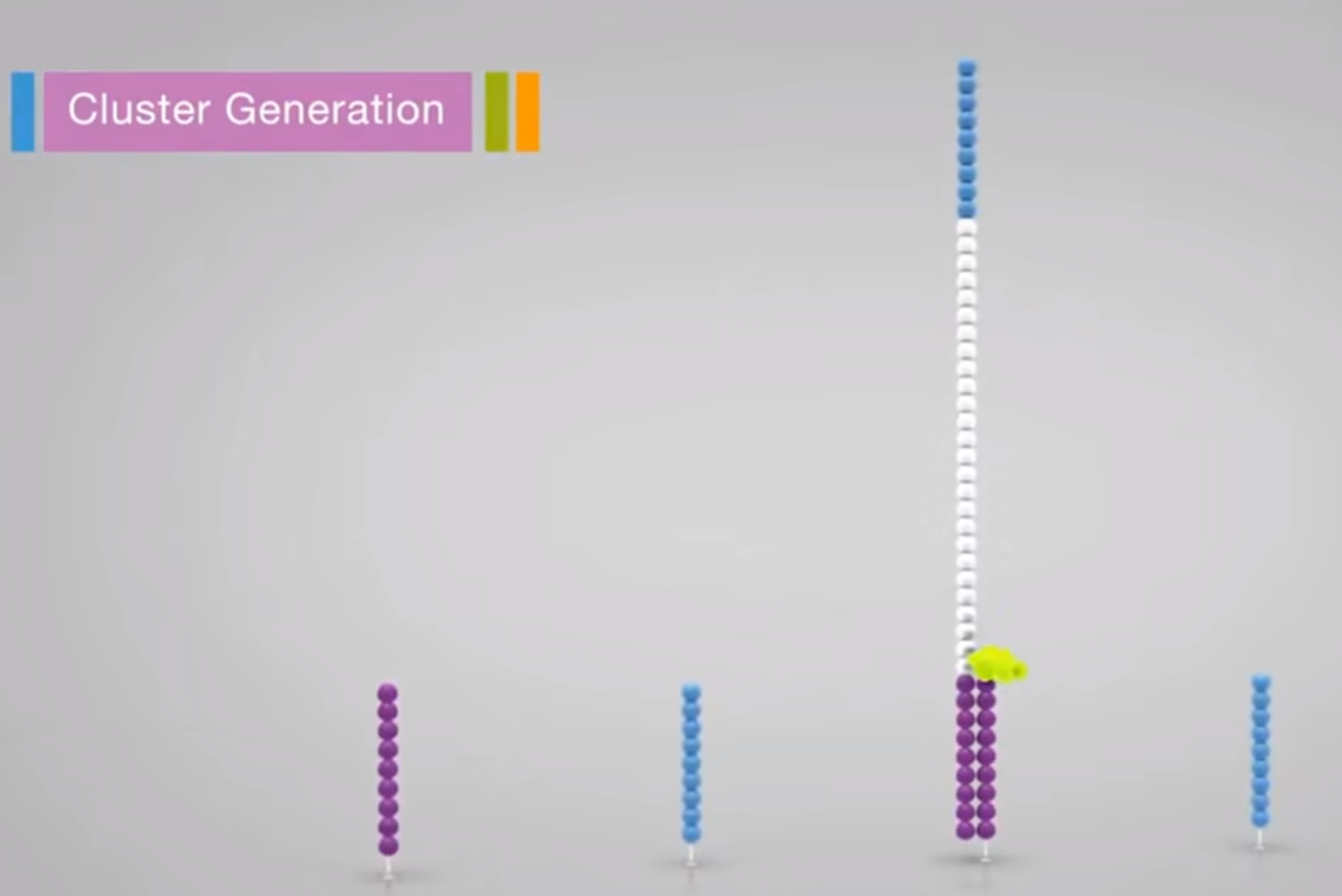
④加入NaOH碱溶液使双链变性后冲洗掉未被固定的单链(即模板链),而反向互补链则在被固定处延伸
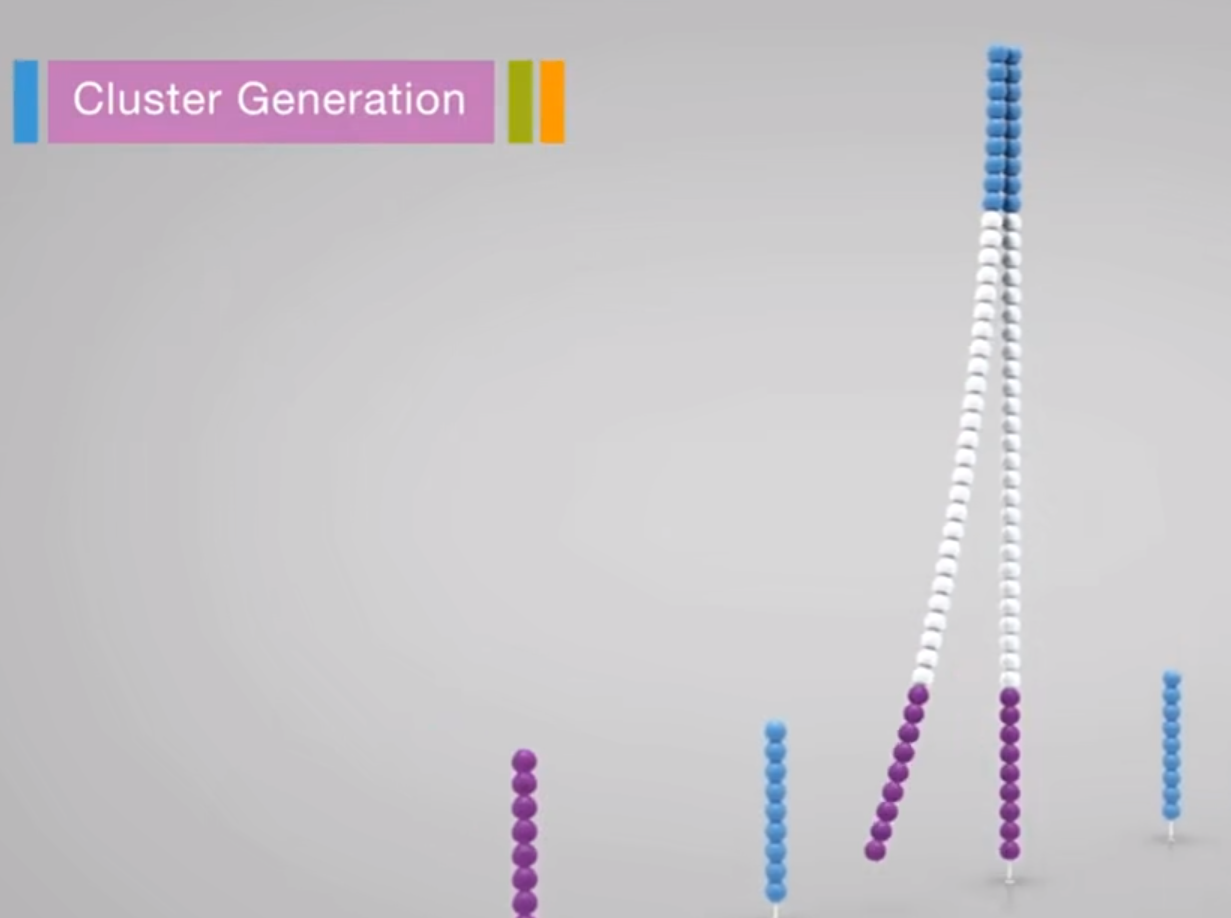
⑤剩下的链在中性溶液中折叠导致另一接头与被固定的对应寡核苷酸片段(作用同上)结合,同样进行复制延长,该过程称为桥式PCR

⑥利用NaOH碱溶液使其发生变性,我们得到两条被固定的反向互补链,其中紫色底座是被测序列的反向互补,蓝色底座的是正链
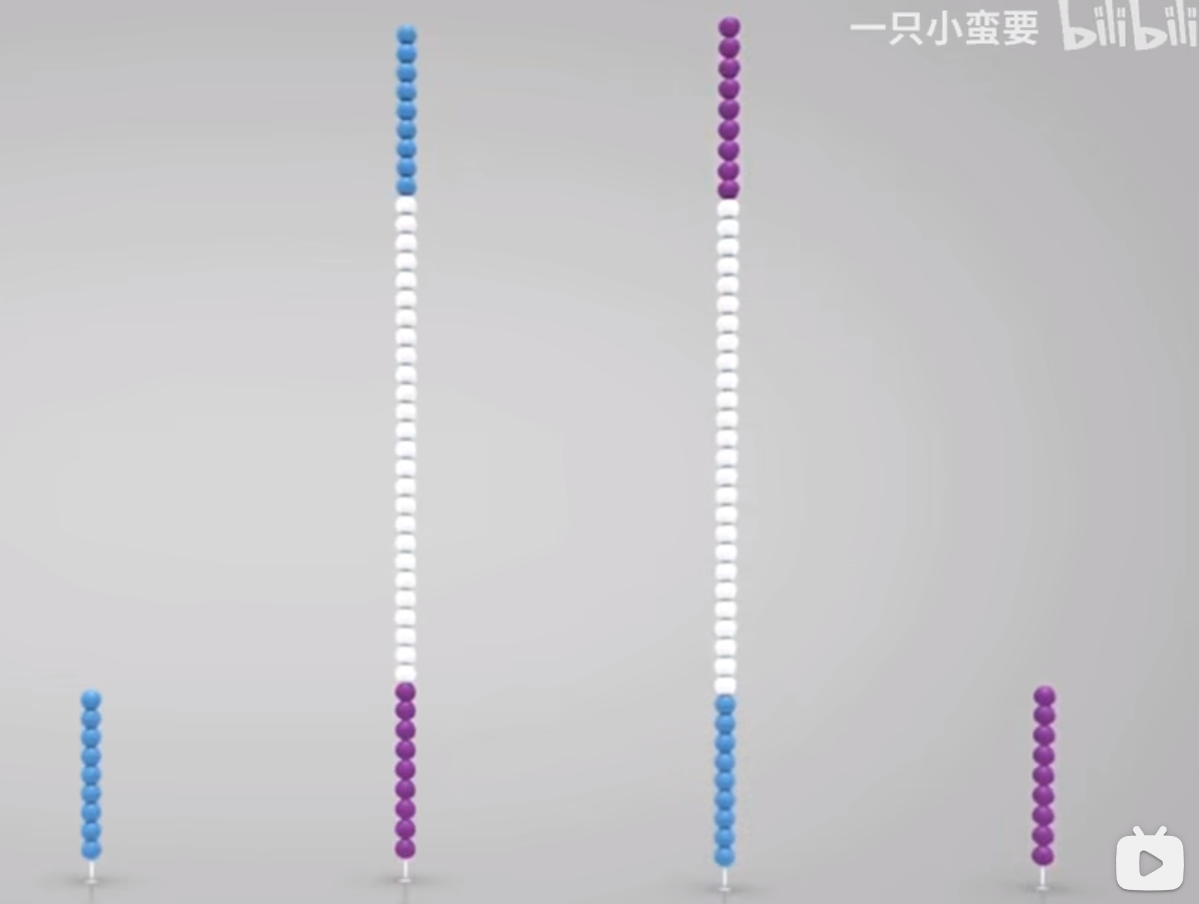
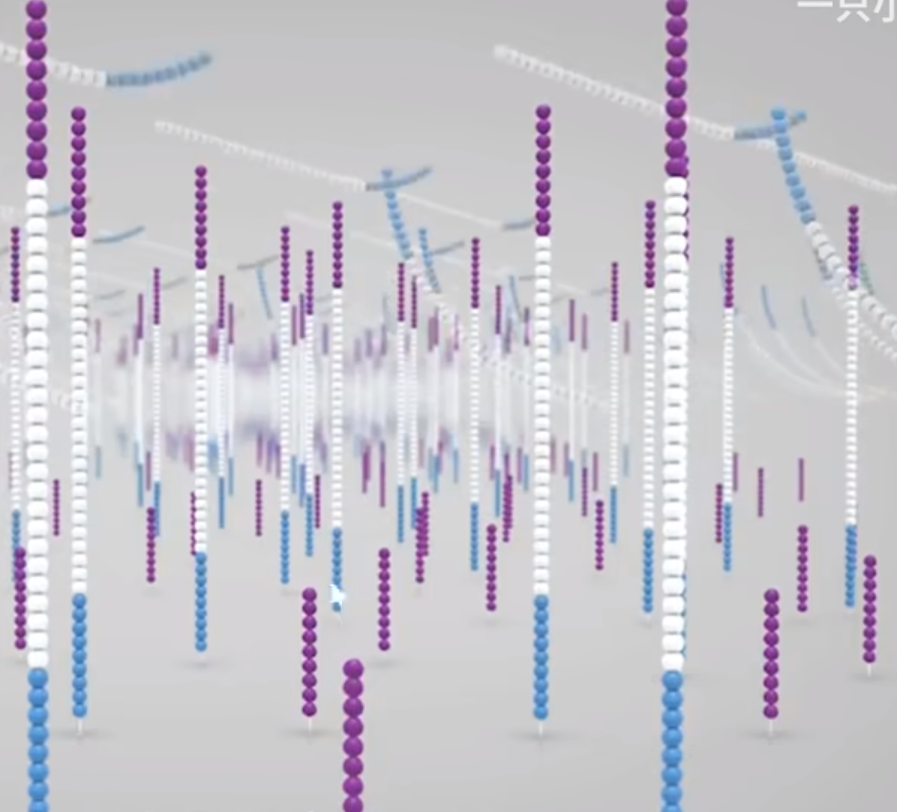
⑦多次重复上述过程,我们得到许多相邻的核苷酸链被固定在lane上,并且这些核苷酸链都来自同一个片段的正反链

⑧将反向互补链引物上的一个基团用酶切除,用碱溶液清洗。此时只剩下正链被固定,这些被固定的正链称为簇,一定区域内只包含这些相同的单链。簇在下一步的测序中可以将荧光信号放大。
1.3边合成边测序
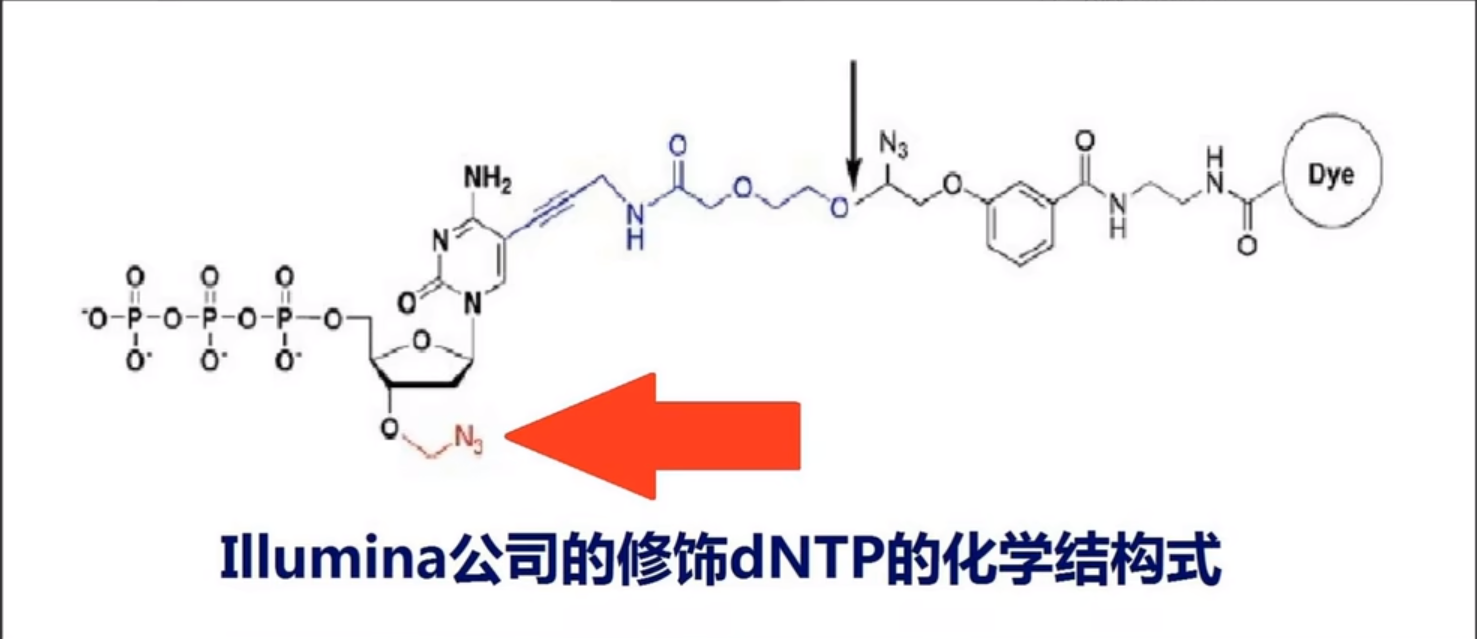
①单端测序:加入对应的引物P5和含有可逆荧光基团的dNTP在酶的作用下对应互补,不同的dNTP对应不同的颜色,使簇表现出不同的颜色并被拍摄识别。但是在叠氮基团和荧光基团存在的情况下无羟基进行延伸,因此需要使用化学试剂切除该基团使其3’端变回羟基,从而加入下一个携带可逆荧光基团的dNTP。如此往复可以实现测序,循环的次数取决于reads的长度。并且该过程是flowcell中数以亿计的簇同时进行。

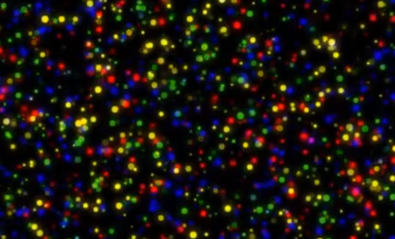
在第一次读段完成后,将上一步读段产生的双链清洗去除,加入index1对应的引物对barcode进行测序,获得簇对应的样本。
②双端测序:将上一步的正向链进行桥式PCR,获得被固定的负链,再用另一个引物P7进行负链的测序。这个过程的意义在于:有些reads长度很长,超出了illumina单端测序的范围,超过的部分其准确性将严重降低,可靠度下降,故采用双端测序的方式弥补测序片段长度。需要注意的是,P7引物下测序所得的序列是反向互补链为模板测得的。
1.4数据分析
通过barcode将数百万个reads进行分类,同时给出对应的数据质量评分,保存在fastq文件中。随后我们可以利用这些测序文件进行序列的比对,组装,等具体的研究用途。
视频链接:【中英双语】Illumina测序原理详解 | 边合成边测序/Sequencing by Synthesis_哔哩哔哩_bilibili
二、SoLiD测序
1. 基本流程
SOLiD 的全称为:Sequencing by Oligo Ligation Detection,是ABI公司与2007年推出的测序平台。SOLiD测序平台以四色荧光标记寡核苷酸的连续合成为基础,取代了传统的聚合酶连接反应。
第二代DNA测序,基本上都包括如下三个步骤:
①DNA建库
②DNA扩增
③DNA测序
所以,我们从这三个步骤讲一下SOLiD测序技术。
1.1 DNA 建库
首先将DNA分子进行打断,所有的二代测序平台所等测定的片段长度有限,所以制定的是片段化的文库。DNA打断一般采用酶切打断或者物理打断(比如超声波打断)。
SOLiD支持两种文库:
①片段文库:
是在打断的DNA分子两端,连接接头,在SOLiD中,我们称之为P1和P2接头。

②末端匹配文库:
在打断的DNA分子两侧连接上一个中间接头,再把连接上中间接头的序列进行环化,成为一个环状分子
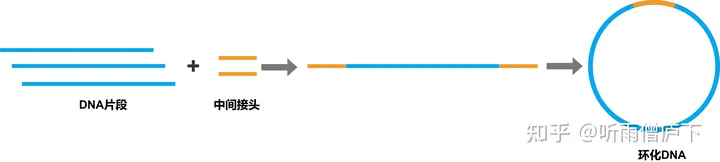
环化后的DNA分子,通过EcoP15进行酶切,特意切出具有27bp的片段,然后只收取具有中间接头的序列。然后再在该片段两端连接P1和P2。过程如下图:
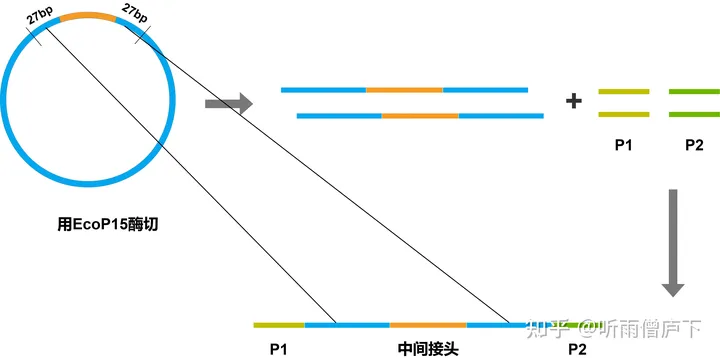
这种方式的片段长度有限,也有其他的方式可以切出更长的片段,因为原理类似,本文不做详细探讨。
1.2 DNA扩增
扩增的目的是将DNA片段复制足够多,从而为后续测序提供足够的样片。
首先,将DNA片段固定在微珠上,然后和矿物质油混合并高速震荡,形成“油包水”的乳液环境,通过一系列方法,拿到“一液滴,一磁珠,一模板”的形式。这种方式最大特点是,可以形成数目庞大的独立反应空间以进行DNA扩增。
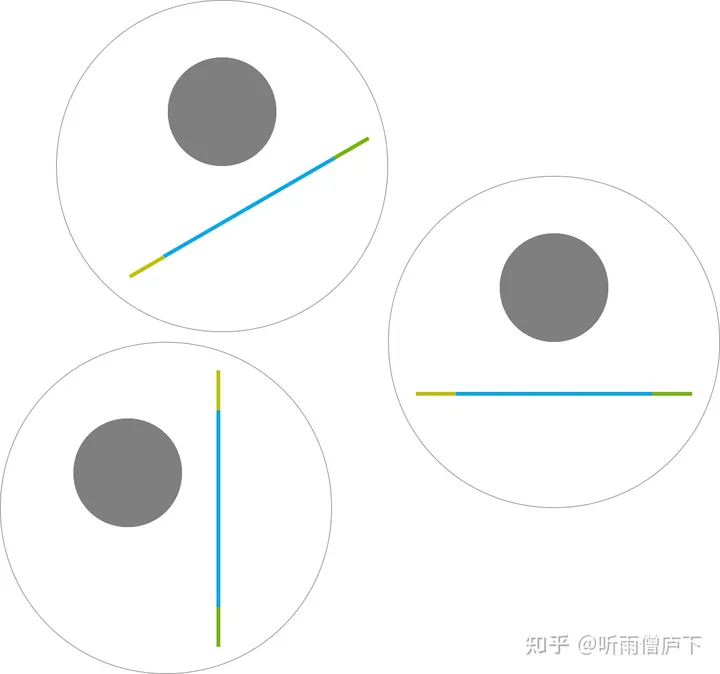
在每个液滴中,进行PCR扩增,每个微珠都会产生数十万条单克隆模板。
接下来,打破油水结构,可以使用2-丁醇打破油水分享。并通过分子杂交和差速离心的方法对模板磁珠进行收集,得到最终的模板磁珠产物。
最后,将含有DNA模板的磁珠结合在SOLiD玻片表面。磁珠是SOLiD测序的最小单元。每个磁珠SOLiD测序后形成一条序列。
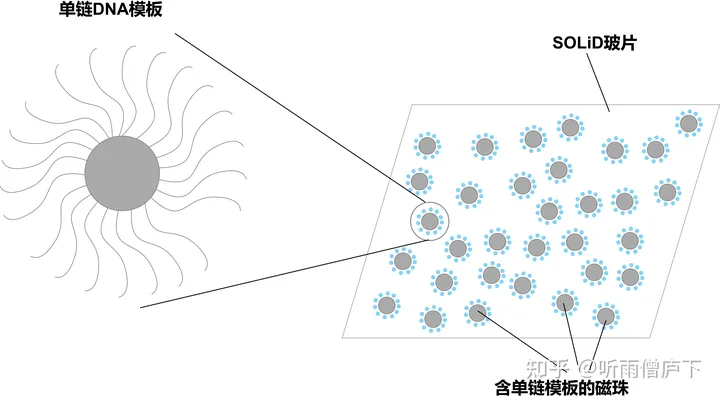
1.3 DNA测序
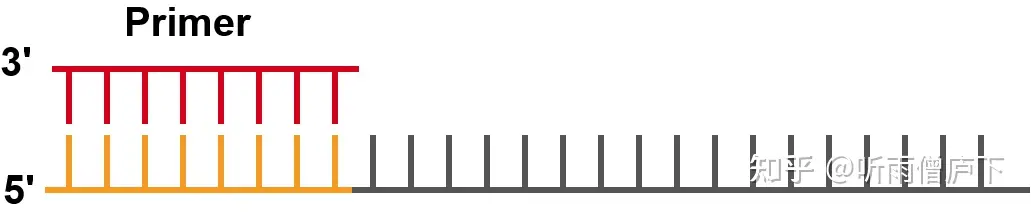
将测序引物(primer)和模板链结合,加入引物是因为DNA聚合酶不能重头复制需要前面有一小段起点。
连接探针序列,探针序列使用一个含有八个碱基的特殊序列,探针3’ 端的1、2 位构成的碱基对,表征探针染料类型;35位的“n”为随机碱基;68位的“z”是可以和任何碱基配对的特殊碱基。如下图。
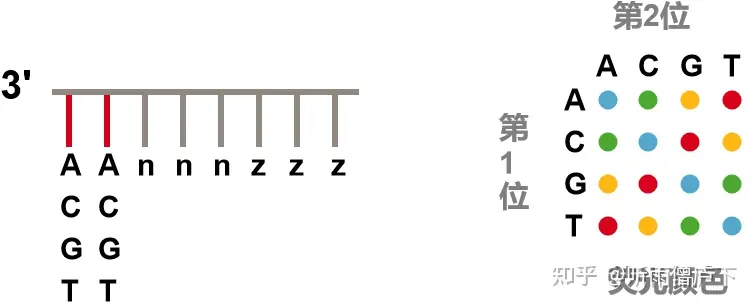
单向SOLiD测序包含五次测序反应,每一次测序反应会连接15位的碱基,切除68位的碱基,同时记录下荧光颜色,荧光颜色由1~2位碱基决定,如图8右。这样,8个碱基的探针最后只有5个碱基连接到模板上去。
测序反应步骤如下:
- 投入一种8碱基荧光探针。测序仪记录下第1、2位碱基代表的颜色信息。去除第6~8位碱基,为下一次连接反应做准备。
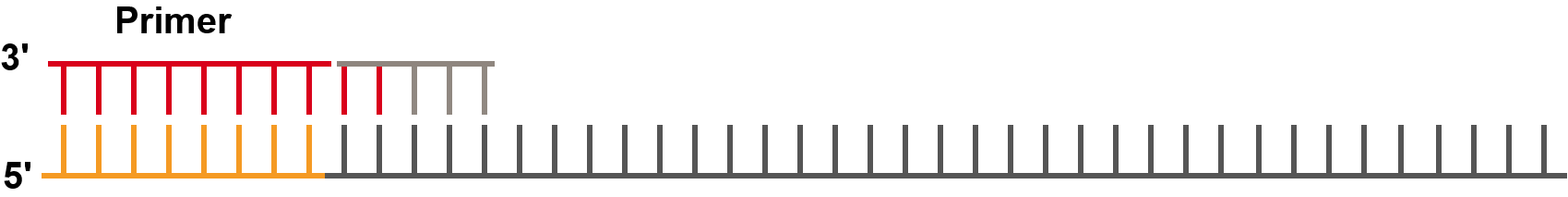
- 重复第一步,共5次。
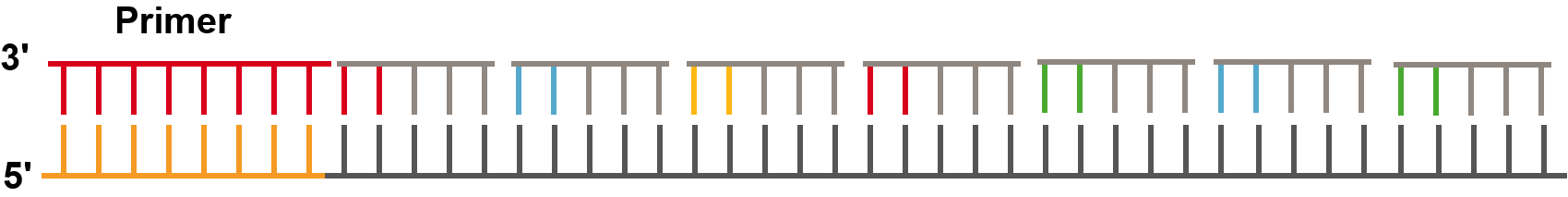
- 接下来重置引物,这个引物和第一次的引物不同,在接头上经过一次退位,长度为n-1(第一轮引物长度是n)。这样和第一轮相比,测序有了一个碱基的错位。
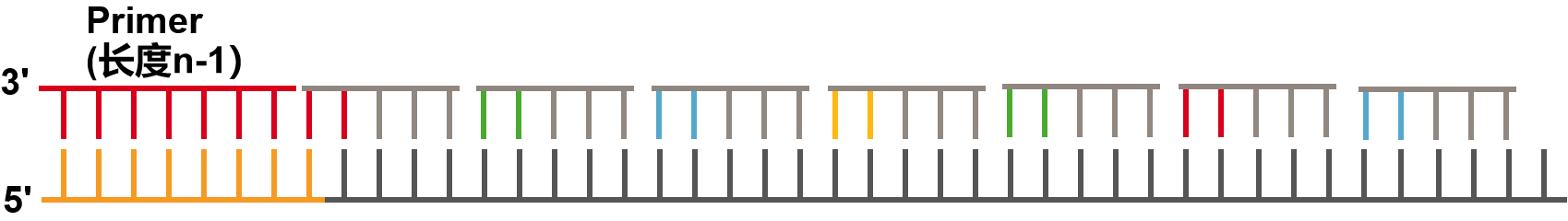
重置,引物长度变为n-2。
重置,引物长度变为n-3。
重置,引物长度变为n-4。这样最后,每位碱基都经过了两轮测试。如下表。
| 轮次 | 引物长度 | 第一次 | 第二次 | 第三次 | 第四次 | 第五次 |
|---|---|---|---|---|---|---|
| 1 | n | 1,2 | 6,7 | 11,12 | 16,17 | 21,22 |
| 2 | n-1 | 0,1 | 5,6 | 10,11 | 15,16 | 20,21 |
| 3 | n-2 | 4,5 | 9,10 | 14,15 | 19,20 | 24,25 |
| 4 | n-3 | 3,4 | 8,9 | 13,14 | 18,19 | 23,24 |
| 5 | n-4 | 2,3 | 7,8 | 12,13, | 17,18 | 22,23 |
然后,通过荧光颜色进行解码,确定碱基序列。由于0号位是一个已知序列,那么根据0,1号位的颜色,第1号位就能推得。推得1号位以后,再找到第1,2号位的颜色,推得2号位。以此类推从而得到整组序列。
2. 总结
由于每个碱基都会测序两次,所以SOLiD是第二代测序平台中精度最高的,然而因为读取长度受限,所以运行速度较慢。
但是,由于市场竞争和公司发展等原因,目前该平台已经淡出市场。
原文链接:第二代DNA测序原理 - SOLiD 测序技术 - 知乎 (zhihu.com)
三、Roche 454测序
1.罗氏 454 测序技术
第二代DNA测序,基本上都包括如下三个步骤:
①DNA建库
②DNA扩增
③DNA测序
所以,我们从这三个步骤讲一下罗氏 454测序技术的。
1.1 DNA建库
因为测序仪器的测序长度有限,所以首先需要将DNA样片打断,打断后的DNA分子具有两个粘性的末端,在这两个末端上分别接上A接头和B接头。
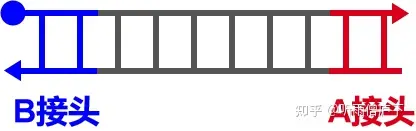
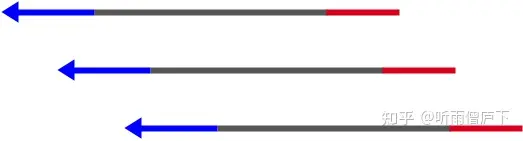
当然,在这个过程中,也会出现两边都是A接头或者两边都是B接头的情况,这种情况会通过一些手段进行分离(磁珠分离的方法,这里不具体描述)。最终,我们得到了一批具有AB接头的单链DNA片段。
1.2 DNA扩增
扩增的目的是将DNA片段复制足够多,从而为后续测序提供足够的样片。
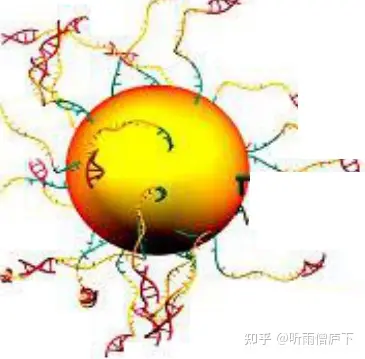
将DNA片段固定在微珠上,然后和矿物油混合并高速震荡。获得了“油包水”的乳液环境。在理想的乳液中,每一个液滴中,仅含有一个微珠和一条模板,如图3。但事实上,也会存在其他三种情况:1. 仅有微珠;2. 有微珠却有多条模板;3.仅有模板 这几种其他情况会通过一定的方法洗脱,这里就不具体展开。
然后进行PCR扩增,每个微珠都将生产数十万条固定的单克隆模板。
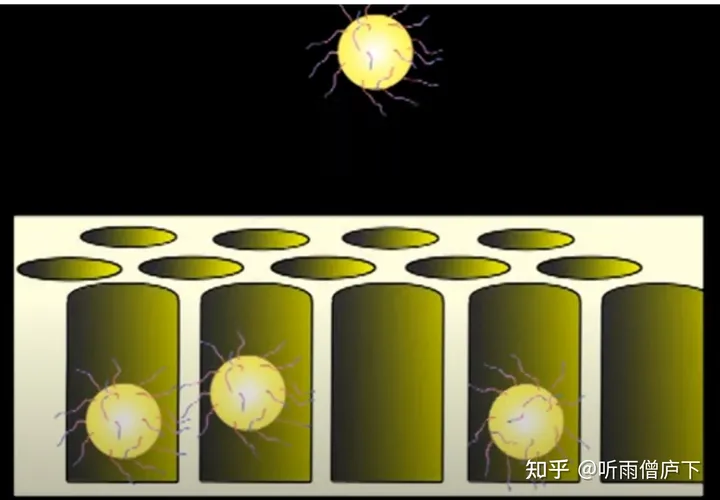
然后收集微珠,将微珠加入芯片中。这里的芯片是一块具有多孔结构的高分子板。每个孔的孔径略大于微珠的直径,以保证一个孔中只能存在一个微珠。每一个孔我们都可以看做一个“测序通道”,通过多孔结构,就能实现多通道并行测序。
1.3 DNA测序
454测序法采用的是焦磷酸测序法。简单来说就是每次放入不同的碱基,如果对应上了,就会产生荧光反应,被感光元件接收到,之前我们通过DNA扩增,将片段复制了几十万份,这样产生的光强度也被放大了几十万倍,这样才能被感光元件捕捉到。因为放入的碱基是已知顺序的,比如A->G->C->T,我们就能得到放入不同碱基的感光信号。这样不断的循环,我们就能拿到一条完整的序列的信号。因为芯片面板上布满了大量的微孔,所以每个微孔都能发生荧光反应,所以,我们拿到的荧光信号是如下图的。将多幅这样的荧光照片进行图像识别,就实现了并行测序。

再将不同的序列进行拼接,我们就能得到了完整的DNA序列。
2. 454 测序法的缺点
刚刚我们了解了454测序法的原理,也就是通过放入不同碱基后所发生的荧光反应,来判断DNA序列。但是,当出现连续相同的碱基序列时,可能会存在错误。举个例子,当DNA链上出现了连续多个A,这样在反应中,就会加上多个T,那么如何判断多少个T呢?只能通过荧光信号的强度来判断,这里就有可能造成结果不准确。所以454的测序错误不是来自核苷酸的替换,而是插入或者缺失。