水稻kozak序列的寻找
Updated on: 2024-03-08 08:08:40
一、kozak序列
Kozak序列是位于真核生物mRNA 5’端帽子结构后面的一段核酸序列,通常是GCCACCAUGG,表达式为“[A/G]NNATGG”。它可以与翻译起始因子结合而介导含有5’帽子结构的mRNA翻译起始,可以认为是RBS。对应于原核生物的SD序列。存在于真核生物mRNA的一段序列,其在翻译的起始中有重要作用。
起始密码子两端序列为:—— G/N-C/N-C/N-ANNAUGG——,如GCCACCAUGG、GCCAUGAUGG时,转录和翻译效率最高,特别是-3位的A对翻译效率非常重要。该序列被后人称为Kozak序列,并被应用于表达载体的构建中。
kozak规则:
(1)第4位的偏好碱基为G;
(2)AUG的5’端约15bp范围的侧翼序列内不含碱基T;
(3)在-3,-6和-9位置,G是偏好碱基;
(4)除-3,-6和-9位,在整个侧翼序列区,C是偏好碱基。
Kozak规则是基于已知数据的统计结果,不见得必须全部满足,一般来说,满足前两项即可
二、水稻cDNA下载
在ensemble plant数据库下载:Index of /pub/plants/release-58/fasta/oryza_sativa (ebi.ac.uk)
三、源码
本代码实现的是在水稻完整的cDNA序列中,寻找每个起始密码子周围的kozak序列的数量。
思路:先将FASTA文件利用bio库分成一个一个基因,随后在每个基因中找到所有的ATG,并对这些ATG周围的7个bp进行判断是否符合kozak序列规则,若满足则再寻找下游3bp倍数上最近的终止密码子,若这个终止密码子与ATG的距离大于10个aa则认为其编码了一个能折叠的多肽链(现实情况不一定是这样的)。
缺点:这种情况可能出现一个基因有满足条件的多个kozak序列————————是否存在同一个基因在翻译水平上的调控,即一个长的ORF可以翻译出两个一长一短的多肽链。
这个缺点可以通过加一步筛选更长的肽链来弥补,但是我认为这种两个kozak序列对应一个终止密码子的情况是需要生物学验证的。
from Bio import SeqIO
import re
import pandas as pd
import numpy as np
Gene = []
ture_kozak_num = []
candidate_kozak_num = []
Kozak_sequence = []
candidate_kozak = 0
ture_kozak = 0
# 打开cDNA文件
with open("Oryza_sativa.IRGSP-1.0.cdna.all.fa", "r") as handle:
# 逐个处理每个基因的序列
for record in SeqIO.parse(handle, "fasta"):
sequence = str(record.seq)
Gene.append(record.id)
# 寻找序列中所有ATG序列的索引位置
indexes = []
index = -1
while True:
index = sequence.find("ATG", index + 1) # 从ATG后一位继续检索
indexes.append(index)
if index == -1:
break
for candidate_index in indexes:
# 如果无ATG序列
if candidate_index == -1:
print(f"Gene {record.id}: end\n")
#最终每个基因的candidate_kozak和ture_kozak都会在此计算完成并被加入到列表中
candidate_kozak_num.append(candidate_kozak)
ture_kozak_num.append(ture_kozak)
candidate_kozak = 0
ture_kozak = 0
# 如果ATG是第一个碱基,无法构成kozak序列,这种情况可能是测序遗漏了前面几个bp,也可能恰好是ATG的utr区,无法判断
elif candidate_index == 0:
print(f"Gene {record.id}: A ATG initio ")
# 如果含有ATG序列且前面有3个碱基足够构成kozak序列
elif candidate_index >= 3 and candidate_index + 4 <= len(sequence):
# 获取前后各三个碱基的7个bp片段
kozak_ATG_codon = sequence[candidate_index - 3:candidate_index + 4]
# 定义kozak序列pattern
pattern = re.compile(r"[AG]..ATGG")
# 判断是否满足Kozak序列规则
if re.match(pattern, kozak_ATG_codon):
candidate_kozak += 1
print(f"Gene {record.id} candidate Kozak sequence found: {kozak_ATG_codon},number: {candidate_kozak}")
# 若满足kozak序列规则,判断是否有终止密码子
# 寻找候选kozak序列后所有的终止密码子的索引
terminal_indexes = []
for a in re.finditer(r'TAA|TAG|TGA', sequence[candidate_index:]):
terminal_indexes.append(a.start())
#所有在3的倍数位置的终止子索引
correct_terminal_index = [i for i in terminal_indexes if i % 3 == 0]
#最近的在3的倍数位置的终止子索引
if correct_terminal_index:
nearest_terminal_index = min(correct_terminal_index)
#至少间隔有100个氨基酸就认为是正确的终止子
if nearest_terminal_index >= 300:
ture_kozak += 1
print(f"Gene {record.id}:true Kozak sequence found: {ture_kozak}")
else:
print(f"Gene {record.id}: No terminal codon found")
# for code in re.finditer(re.compile(r'TAA|TAG|TGA'), sequence):
# terminal_code_indexes = code.start()
# # 判断是否终止子在3的倍数位置,且间隔至少有十个氨基酸就认为是正确的终止子
# if (terminal_code_indexes-candidate_index) % 3 == 0 and terminal_code_indexes-candidate_index >= 300:
# ture_kozak += 1
# print(f"Gene {record.id}:true Kozak sequence found: {ture_kozak}")
# break
elif candidate_index + 4 >= len(sequence):
# ATG在末尾的,也不排除测序错误遗漏了后半段的编码区,但是可能性非常小,因为CDS很长
print(f"Gene {record.id}: A ATG tail")
df = pd.DataFrame()
df.insert(0, 'Gene', Gene)
df.insert(1, 'ture_kozak', ture_kozak_num)
df.insert(2, 'candidate_kozak', candidate_kozak_num)
#输出df中gene为Os02t0788600-01的数据
print(df[df['Gene'] == 'Os02t0788600-01'])
print(df)
# df.to_csv('kozak.csv', index=False)
最终会生成一个csv文件统计对应了终止密码子的kozak序列和未对应终止密码子的kozak序列。
有了上述思路后,又做了一个优化版代码,大大提高运行效率。优化在于减少了很多不必要的变量,大大提高了可读性。
import re
import pandas as pd
from Bio import SeqIO
def find_kozak_sequences(record):
gene = record.id
sequence = str(record.seq)
candidate_kozak_num = 0
true_kozak_num = 0
for match in re.finditer(r'ATG', sequence):
start_index = match.start()
# 检查 ATG 前后是否有足够的序列
if start_index >= 3 and start_index + 4 < len(sequence):
kozak_sequence = sequence[start_index - 3:start_index + 4]
if re.match(r'[AG]..ATGG', kozak_sequence):
candidate_kozak_num += 1
# 检查附近的终止密码子
stop_codon_indices = [m.start() for m in re.finditer(r'TAA|TAG|TGA', sequence[start_index:])]
nearest_stop_index = min(filter(lambda x: x % 3 == 0 , stop_codon_indices), default=-1)
if nearest_stop_index >= 150:
true_kozak_num += 1
return gene, candidate_kozak_num, true_kozak_num
if __name__ == "__main__":
records = SeqIO.parse("Oryza_sativa.IRGSP-1.0.cdna.all.fa", "fasta")
results = [find_kozak_sequences(record) for record in records]
df = pd.DataFrame(results, columns=['基因', '候选Kozak', '真Kozak'])
print(df)
df.to_csv('kozak.csv', index=False)
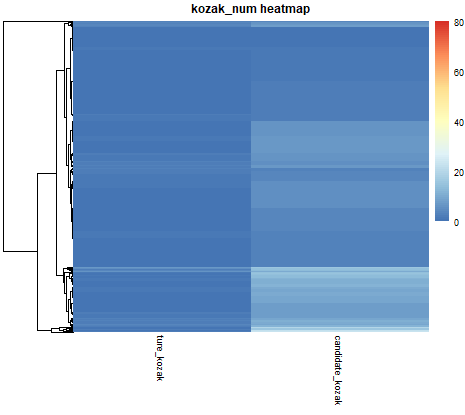
最后采用R语言的pheatmap绘制了一个热图,进行可视化。
library(pheatmap)
library(RColorBrewer)
setwd('E://R语言学习文件/')
dataset <- read.csv("kozak.csv",header = TRUE, row.names= 1)
pheatmap(dataset,#表达数据
#display_numbers = F, # 矩阵的数值是否显示在热图上
#number_format = "%.2f", # 单元格中数值的显示方式
#fontsize_number = 8, # 显示在热图上的矩阵数值的大小,默认为0.8*fontsize
cluster_rows = T,#行聚类
cluster_cols = F,#列聚类
#clustering_method = "complete", #表示聚类方法,包括:
#‘ward’, ‘ward.D’, ‘ward.D2’, ‘single’, ‘complete’, ‘average’, ‘mcquitty’, ‘median’, ‘centroid’
show_rownames = F,#显示行名
show_co1names= T,#显示列名
#scale = "column",#对行标准化'none', 'row' or 'column
#color = colorRampPalette(colors = c("blue","white","red"))(100),#颜色
legend = T,#显示图例
#legend_breaks = c(0,1),#图例断点显示
#breaks = c(0,0.45,0.55,1),#图例断点
#legend_labels = c("non-resist","resist"), # 图例断点标注的标题
#cellwidth = 7, cellheight = 4,#调整热图单元格宽度、高度
#treeheight_row = 30, treeheight_col = 18,#调整行列聚类树的高度
fontsize =7, fontsize_row = 7, fontsize_col = 7, #热图中字体大小、行、列名字体大小
main = "kozak_num heatmap", #表示热图的标题名字
#annotation_col = annotation, #添加列注释信息
#annotation_row = annotation_row, #添加行注释信息
annotation_1egend=F,#显示样本分类
#annotation_colors =ann_colors,#设置行注释颜色
#cutree_rows=3,cutree_cols=7,#根据行列的“聚类”将热图分隔开
#gaps_row = c(12, 21),#未进行聚类时手动分割
#gaps_col = c(1,11,15,19,28,43,48),#未进行聚类时手动分割
#labels_row = NULL, #使用行标签代替行名
#labels_col = NULL, #列表签代替列名
)#热图基准颜色
绘制的热图如下: