水稻DRE序列寻找
Updated on: 2024-03-11 17:25:11
1.题目

2.代码
2.1 Linux shell
# 分离染色体并计数每个染色体上序列数目
csplit -f rice_chromosome all.con '/>Chr/' '{*}'
for file in rice_chromosome*; do
chromosome=$(grep -o '>Chr[0-9]*' "$file") # 提取染色体编号
sequence_count=$(grep -o “[AG]CCGC.T" "$file" | wc -l)
echo "$chromosome $sequence_count" >> sequence_counts.txt
done
# 使用 GNU Plot 绘制柱状图
gnuplot << EOF
set terminal pngcairo enhanced font 'Arial,12'
set output "sequence_counts_plot.png"
set title "Sequence Counts on Each Chromosome"
set xlabel "Chromosome"
set ylabel "Sequence Count"
set style fill solid
set boxwidth 0.5
set xtics rotate by -45
plot "sequence_counts.txt" using 2:xtic(1) with boxes lc rgb "blue" notitle
EOF
echo "Plot saved as sequence_counts_plot.png"
2.2 python代码
from Bio import SeqIO
import matplotlib.pyplot as plt
import re
# 读取文件
fasta_file = "all.con"
records = SeqIO.parse(fasta_file, "fasta")
# 统计每个染色体上序列的数量
chromosome_counts = {}
for record in records:
chromosome = record.id.split()[0] # 提取染色体编号
if chromosome not in chromosome_counts:
chromosome_counts[chromosome] = 0
#找到所有符合条件的motif
chromosome_counts[chromosome] +=
len(re.findall(r'[AG]CCGC.T', str(record.seq)))
# 创建柱状图
plt.bar(chromosome_counts.keys(), chromosome_counts.values(), color='blue')
# 添加标题和轴标签
plt.title('Sequence Counts on Each Chromosome')
plt.xlabel('Chromosome')
plt.ylabel('Sequence Count')
# 显示图形
plt.xticks(rotation=-45)
plt.tight_layout()
plt.show()
3. 结果
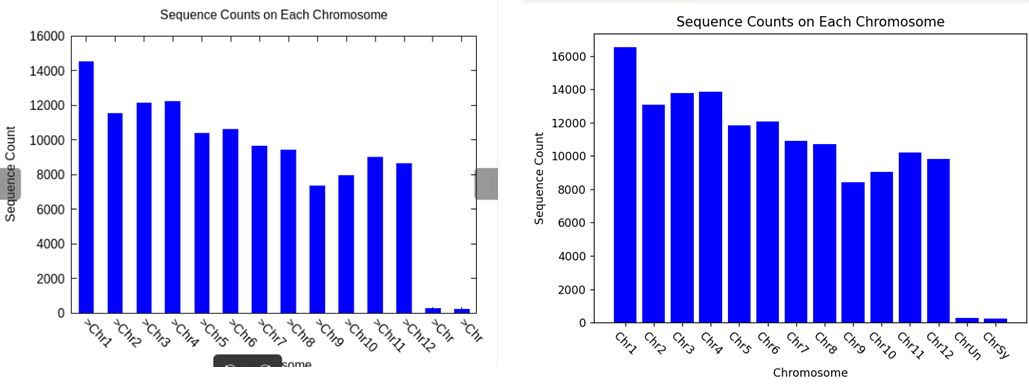
结果的趋势相同,但是Linux脚本的数量在每条染色体上都偏少于python的结果,据此可以怀疑,grep命令无法跨行检索,而源文件all.con确实是如此,但是python的SeqIO.parse()命令可以获取fasta文件的“>序列解释”+下游的所有序列,并且是将序列合并成一条。
4.修改
for file in rice_chromosome*; do
chromosome=$(grep -o '>Chr[0-9]*' "$file") # 提取染色体编号
sequence_count=$(tr -d ‘\n’ < $file | grep -o '[AG]CCGC.T' | wc -l);
echo "$chromosome $sequence_count" >> sequence_counts.txt; done需要注意的是,grep -oc 是返回能够匹配模式的行数,即使加了-o也只能返回行数。因此还是需要wc -l进行计数。
修改后结果如下:
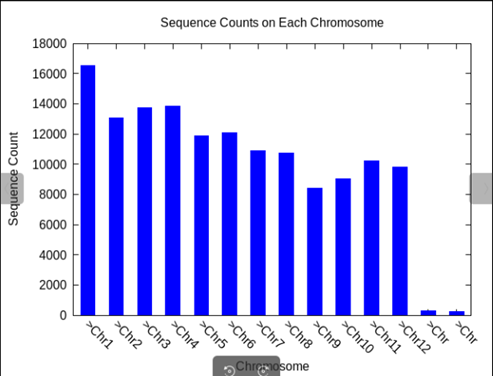
和python结果一样了。